
 English
English Korean
Korean
국내 RA인허가
ICMC교육원

icmcert@naver.com

02-851-3111
체외진단 의료기기 KGMP
체외진단 의료기기 제조·수입 GMP 심사
한국(MFDS) 체외진단 의료기기 GMP
체외진단 의료기기 GMP(Good Manufacturing Practice)
체외진단 의료기기 GMP 제도란 생산∙수입하는 체외진단 의료기기가 안전(Safe)하고, 유효(Effective)하며, 의도된 용도(Intended Use)에 적합한 품질의 제품을 일관성 있게(Consistently) 생산됨을 높은 수준으로 보장하기 위한 품질보증체계를 말한다. 체외진단 의료기기를 생산∙수입하고자 하는 업체는 체외진단 의료기기 설계, 개발, 제조, 시판 후 관리 등 전 과정에 대한 체계적인 품질시스템 확보가 필요하며, GMP 적합인정을 받아야 한다.
체외진단 의료기기 GMP 심사 대상
- 체외진단 의료기기 제조자 또는 수입자
- 임상적 성능시험용 체외진단 의료기기 제조자 또는 수입자
- 적합 인정 및 정기 심사를 받고자 하는 체외진단 의료기기 제조자 또는 수입자
체외진단 의료기기 GMP 심사품목군
품목별 특성에 따라 9개의 GMP 품목군으로 분류하며, 품목군 별로 GMP 적합을 인정한다. (품목군 추가 시 추가 심사필요)
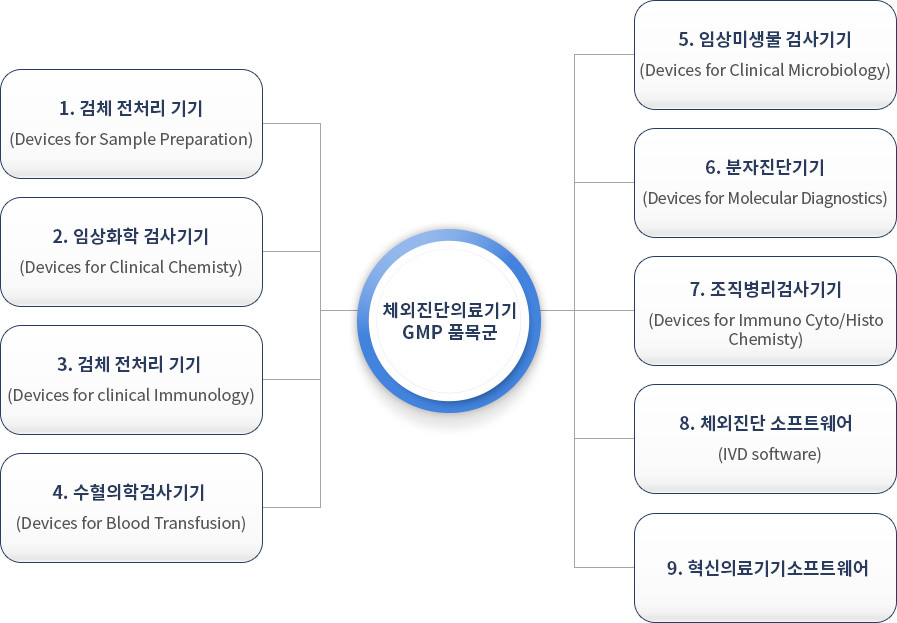
체외진단 의료기기 GMP 심사 구분
- 1
- 최초심사
제조 또는 수입 체외진단 의료기기가 GMP 기준에 적합함을 인정받기 위해 최초로 받는 심사
- 2
- 정기심사
체외진단 의료기기법 시행규칙 및 GMP 고시에 따라 3년마다 1회 받아야 하는 GMP 적용실적에 대한 정기적인 심사 (적합인정서 유효기간 만료 90일 이전에 정기심사 신청)
- 3
- 추가심사
GMP 고시 별표3에 의거 다른 품목군의 체외진단 의료기기를 추가하여 제조 또는 수입하고자 하는 경우 받는 심사
- 4
- 변경심사
제조원의 소재지가 변경(이전)하는 경우 받는 심사. (제품의 품질과 관계가 적은 보관소, 시험실의 변경은 제외)
체외진단 의료기기 GMP 심사 방법
체외진단 의료기기 GMP 심사 주체 (단독: 심사기관, 합동: 지방식약청∙심사기관)
| 구분 | 제조업(국내 제조소) | 수입업(외국 제조소) |
|---|---|---|
2 등급
| 단독 심사 (심사기관 1인) | 단독 심사 (심사기관 1인) |
| 3 · 4 등급 | 합동 심사 (지방식약청 1~2인, 심사기관 1인) | 합동 심사 (지방식약청 1~2인, 심사기관 1인) |
| 심사기간 | 종업원 수에 따라 심사일수(MD) 산정 (2~7일) | 제조공정의 복잡성, 출장일정 (현지 상황 등)을 고려하여 산정 (4~7일) |
체외진단 의료기기 GMP 심사 방법
| 구분 | 제조업(국내 제조소) | 수입업(외국 제조소) |
|---|---|---|
최초심사
| 현장, 서류심사 | 현장, 서류심사 |
| 추가심사 | 서류 심사 | 서류 심사 |
| 변경심사 | 현장, 서류심사 | 현장, 서류심사 |
| 정기심사 | 현장, 서류심사 (1등급 체외진단 의료기기는 정기심사 대상에서 제외) |
현장, 서류심사 (1등급 체외진단 의료기기는 정기심사 대상에서 제외) |
체외진단 의료기기 GMP 심사 신청
체외진단 의료기기 GMP 심사 신청
적합성 인정 등 심사 신청서에 구비서류를 첨부하여 심사기관에 제출
- 체외진단 의료기기 적합성인정 등 심사 신청서
- 체외진단 의료기기 제조(수입)업 허가증 사본 또는 조건부 제조(수입)업 허가증 사본(최초심사 또는 임상적 성능시험용 체외진단 의료기기의 경우 제외)
- 적합성인정 등 심사에 필요한 자료
- 체외진단 의료기기 기술문서 등의 심사결과 통지서 사본(임상적 성능시험용 체외진단 의료기기의 경우 기술문서 등에 관한 자료)
※ 대표품목에 대하여 1개 제조단위 이상의 품질관리 실적 필요 (완제품 시험 성적서 구비)
체외진단 의료기기 GMP 심사 절차
체외진단 의료기기 GMP 심사 절차
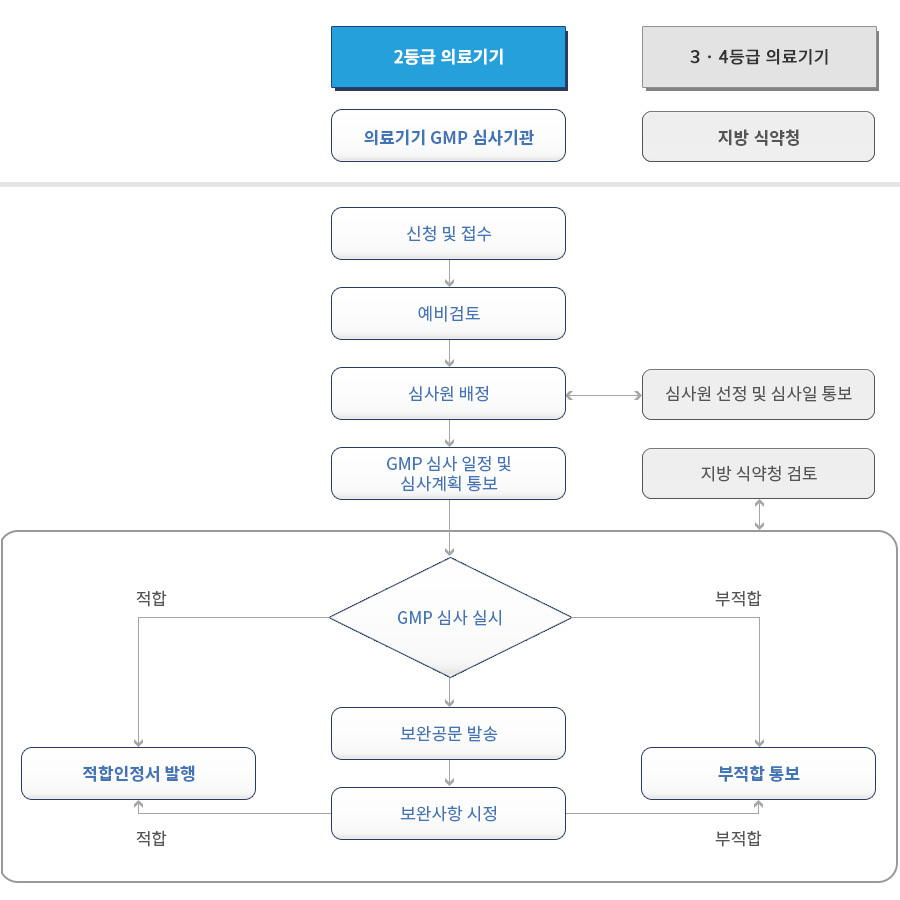
체외진단 의료기기 GMP 평가 기준
-
평가표 평가기준A (적절함)
GMP 기준 요구사항의 준수가 인정되는 경우 B (보완필요)GMP 기준 요구사항을 미행하지 않은 경우
GMP 기준 요구사항을 준수하고 있으나 준수여부의 입증근거, 실현가능성, 기록이 미흡한 경우C (부적절함)“보완필요”에 대해 보완되지 않거나 의료기기법령을 위반한 경우 D (해당없음)GMP 기준의 요구사항에 해당되지 않는 경우 -
적합성 판정기준적합
심사 기준 별 심사결과, 모든 항목이 적절(A)한 경우 보완심사 기준 별 심사결과, 1개 이상의 항목이 보완필요(B)인 경우 부적합심사 기준 별 심사결과, 1개 이상의 항목이 부적절(C)한 경우
체외진단 의료기기 GMP 요구사항
체외진단 의료기기 GMP 프로세스
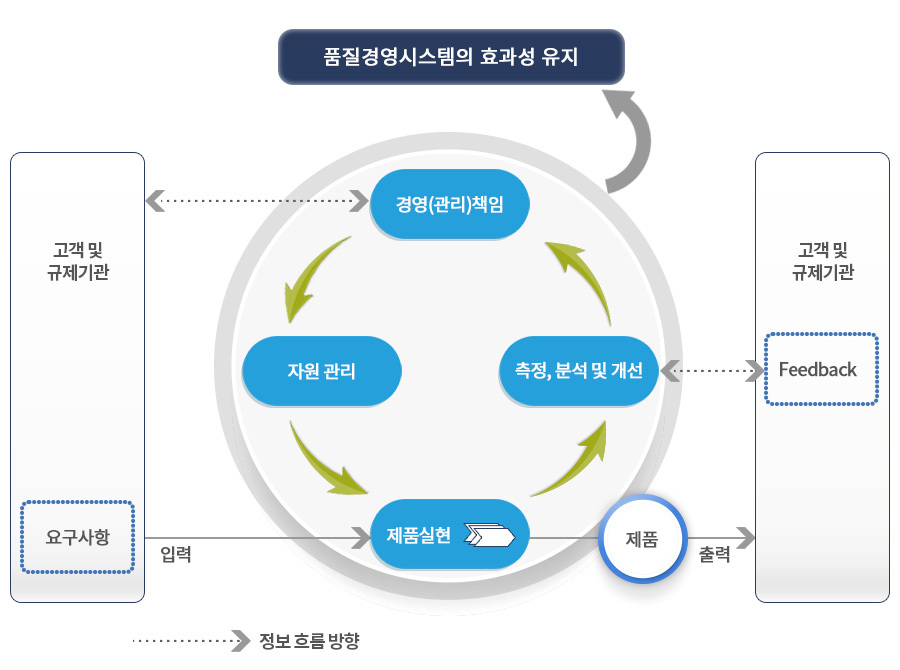
체외진단 의료기기 GMP 요구사항
| 기준 | 세부 요구사항 | 기준 | 세부 요구사항 |
|---|---|---|---|
4. 품질경영시스템
| 4.1 일반 요구사항 |
7. 제품실현 | 7.1 제품실현의 기획 |
| 5. 경영책임 | 5.1 경영의지 |
8. 측정, 분석 및 개선 | 8.1 일반 요구사항 |
| 6. 자원관리 | 6.1 자원의 제공 |
체외진단 의료기기 사용적합성 GMP 심사 기준 적용
사용적합성(usability)이란 제품 사용 시 발생할 수 있는 사용오류 등의 위험을 줄이고 사용자가 체외진단 의료기기를 정확하고 안전하게 사용할 수 있도록 설계한 디자인(예: 버튼, 사용자 화면, 사용설명서 등) 특징을 말한다.
체외진단 의료기기 등급별 단계적으로 GMP 사용적합성 적용을 시행하고 있으며, 체외진단 의료기기의 설계 및 개발 등을 수행할 때 사용적합성을 적용하는 품질관리기법을 구체적으로 제시하여 체외진단 의료기기 제조자의 품질관리시스템에 적용해야 한다.
체외진단 의료기기 사용적합성 설계·평가는 ①사용 사양서 준비, ②사용자 인터페이스의 안전성 점검, ③예측 가능한 위해 파악, ④위해 관련 시나리오 작성·선택, ⑤사용자 인터페이스 사양서 수립, ⑥사용자 인터페이스 설계·구현·평가, ⑦사용자 인터페이스 사용적합성 총괄평가 등의 단계로 이루어진다.
2021년 1월 1일부터 2022년 7월 1일까지 등급별로 단계적 시행
| 등급 | 4등급 | 3등급 | 2등급 | 1등급 |
|---|---|---|---|---|
시행일
| 2021. 1. 1 |
2021. 7. 1 |
2022. 1. 1 |
2022. 7. 1 |
체외진단 의료기기 표준코드(UDI) 제도 도입
체외진단 의료기기의 제조∙수입업자는 체외진단 의료기기 용기나 외장에 표준코드(표준화된 체계에 따라 표기되는 숫자, 전자태그를 포함한 바코드 등)를 표시해야 한다.
2019년 7월 1일부터 2022년 7월 1일까지 등급별로 단계적 시행
| 등급 | 4등급 | 3등급 | 2등급 | 1등급 |
|---|---|---|---|---|
시행일
| 2019. 7. 1 |
2020. 7. 1 |
2021. 7. 1 |
2022. 7. 1 |
체외진단 의료기기 사후관리
체외진단 의료기기 실적보고
체외진단 의료기기의 제조∙수입∙수리업자는 매년 1월 1일부터 1월 31일까지 연 1회 전년도 생산 및 수출·수입·수리실적을 [인터넷실적보고시스템]에서 작성하여 제출해야한다.
체외진단 의료기기 공급보고
체외진단 의료기기의 제조∙수입∙판매∙임대업자는 의료기관 판매임대업자 등에 체외진단 의료기기 공급 시 공급한 달을 기준으로 그 다음달 말일까지 [공급내역보고서]를 [의료기기통합정보시스템]을 통해 식약처장에게 제출해야 한다.
2020년 7월 1일부터 2023년 7월 1일까지 등급별로 단계적 시행
| 등급 | 4등급 | 3등급 | 2등급 | 1등급 |
|---|---|---|---|---|
시행일
| 2020. 7. 1 |
2021. 7. 1 |
2022. 7. 1 |
2023. 7. 1 |
체외진단 의료기기 이물보고
체외진단 의료기기 취급자는 체외진단 의료기기 내부나 용기 또는 포장에서 정상적으로 사용된 원재료가 아닌 것으로 사용 시 위해가 발생할 우려가 있거나 사용하기에 부적합한 물질(이물)을 발견한 경우에는 [의료기기 전자민원창구]를 통해 [이물 발견 보고서]를 즉시 식약처장에게 제출해야 한다.
체외진단 의료기기 이상사례 보고
체외진단 의료기기 취급자는 체외진단 의료기기 사용 중 사망 또는 심각한 부작용이 발생하였거나 발생할 우려가 있음을 인지한 경우 그 사실을 안 날로부터 다음에서 정하는 날까지 [의료기기 전자민원창구]를 통해 [의료기기 이상사례 보고서]를 식약처장에게 제출해야 한다.
| 보고대상 | 보고기한 | |
|---|---|---|
체외진단 의료기기취급자 (체외진단 의료기기 제조∙수입∙수리∙판매∙임대업자, 의료기관개설자, 동물병원 개설자) |
7일 이내 |
- 사망이나 생명에 위협을 주는 이상사례를 초래한 경우 ※ 상세한 내용을 최초 보고일 부터 8일 이내에 추가로 보고 |
15일 이내 |
- 입원 또는 입원기간의 연장이 필요한 경우 - 회복이 불가능하거나 심각한 불구 또는 기능 저하를 초래하는 경우 - 선천적 기형 또는 이상을 초래하는 경우 |
|
30일 이내 |
- 기타 중대한 정보 또는 그 밖에 이상사례로서 식약처장이 보고를 지시한 경우 - 외국 정부의 발표 등 조치사항의 경우 ※ 다만 회수계획을 보고한 경우에는 생략 가능 |
|
의료인, 환자, 체외진단 의료기기소비자 |
- 이상사례를 알게된 경우 | |
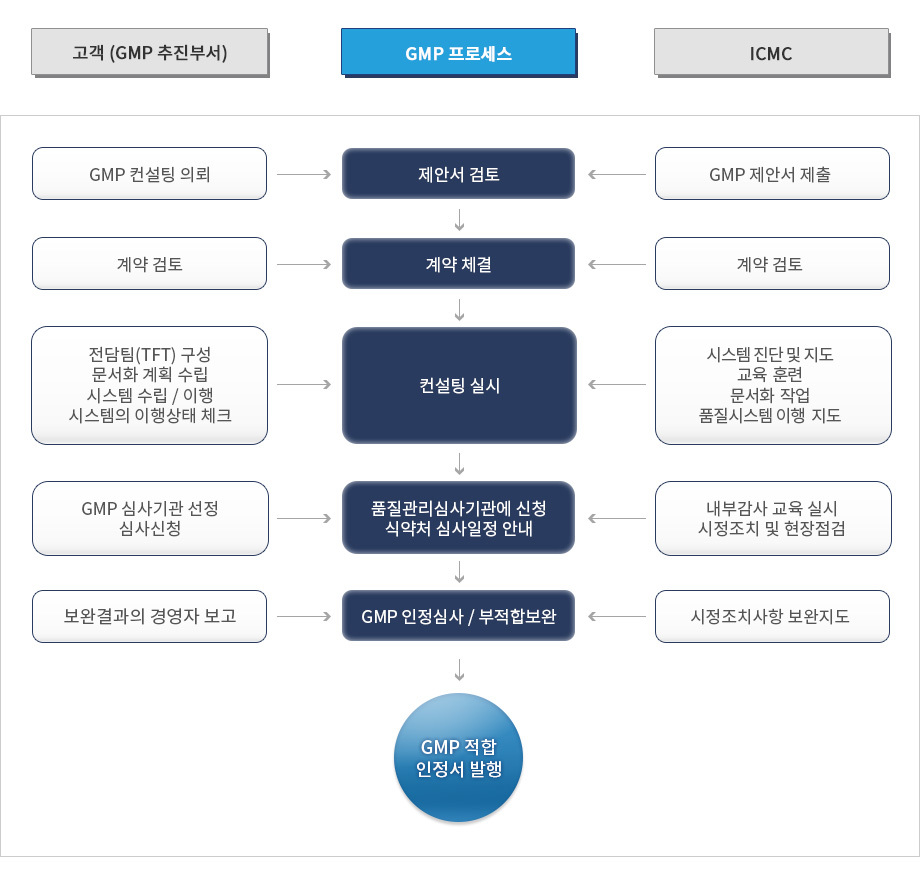
한국(MFDS) 체외진단 의료기기 GMP 컨설팅 추진단계

 10004 certified
10004 certified





