
 English
English Korean
Korean
국내 RA인허가
ICMC교육원

icmcert@naver.com

02-851-3111
체외진단 의료기기 제조∙수입
한국(MFDS) 체외진단 의료기기 법
국내의 체외진단 의료기기 법은 체외진단 의료기기 정의 및 허가 등을 규정하였으며 시행령, 시행규칙으로 세분화하여 아래와 같은 세부내용을 담고 있습니다.
한국(MFDS) 체외진단 의료기기 정의
사람이나 동물로부터 유래하는 검체를 체외에서 검사하기 위하여 단독 또는 조합하여 사용되는 시약, 대조ㆍ보정 물질, 기구ㆍ기계ㆍ장치, 소프트웨어 등 「의료기기법」 제2조 제1항에 따른 의료기기로서 다음 각 목의 어느 하나에 해당하는 제품 (체외진단 의료기기법 제2조)
- 생리학적 또는 병리학적 상태를 진단할 목적으로 사용되는 제품
- 질병의 소인(素因)을 판단하거나 질병의 예후를 관찰하기 위한 목적으로 사용되는 제품
- 선천적인 장애에 대한 정보 제공을 목적으로 사용되는 제품
- 혈액, 조직 등을 다른 사람에게 수혈하거나 이식하고자 할 때 안전성 및 적합성 판단에 필요한 정보 제공을 목적으로 사용되는 제품
- 치료 반응 및 치료 결과를 예측하기 위한 목적으로 사용되는 제품
- 치료 방법을 결정하거나 치료 효과 또는 부작용을 모니터링하기 위한 목적으로 사용되는 제품
한국(MFDS) 체외진단 의료기기 등급 분류
체외진단 의료기기는 사용 목적과 개인 및 공중보건에 미치는 잠재적 위해성의 정도에 따라 4개의 등급으로 분류합니다.
* 체외진단 의료기기 품목 및 품목별 등급에 관한규정 (고시 제 2020-34호)
- 1등급 - 개인과 공중보건에 미치는 잠재적 위해성이 낮은 시약, 법용진단목적으로 사용되는 기기
- 2등급 - 개인에게 중증도의 잠재적 위해성을 가지나 공중보건에 미치는 잠재적 위해성은 낮은 기기
- 3등급 - 진단, 처치, 질병단계 결정 및 치료에 결정적인 영향을 미치는 검사에 시용되는 기기
- 4등급 - 타인에게 수혈이나 이식을 위하여 공여자를 선별하는 검사 혹은 개인위해도가 높은 경우에 사용되는 기기
한국(MFDS) 체외진단 의료기기 품목분류
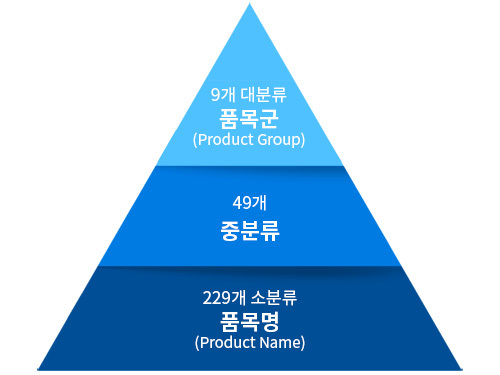
| 체외진단의료기기 품목분류 (대분류별 중분류) | |||
|---|---|---|---|
1. (I) 검체전처리 기기 |
2. (J) 임상화학 검사기기 |
3. (K) 면역 검사기기 |
4. (L) 수혈의학 검사기기 |
- 원심분리장비 |
- 임상화학분석장비 |
- 면역반응검사장비 |
- 수혈의학검사장비 |
5. (M) |
6. (N) |
7. (O) |
8. (P) |
- 임상미생물 검사장비 |
- 분자유전검사장비 |
- 세포 및 조직병리 검사장비 |
- 체외진단 소프트웨어 |
9. 혁신의료기기소프트웨어 |
|||
한국(MFDS) 체외진단 의료기기 인허가 제도
시판 전
시판 전 규제의 주체는 제조자로서 시장 진입을 위한 제품의 안전성과 유효성을 평가하는 분석적 성능시험, 임상적 성능시험, 품목신고, 인증 및 허가, 제조 시스템 관리인 GMP 적합성 인정으로 구분하여 관리되고 있습니다.
시판 후
시판 후 규제는 광고사전심의제도, 부작용/안전성 보고, 재평가 · 재심사 등의 제도를 통해 사후관리가 이루어지고 있습니다.
한국(MFDS) 체외진단 의료기기 인허가 절차
1등급 : 품목 신고 (한국의료기기안전정보원, NIDS)
- 품목신고 신청서 [체외진단 의료기기 전자 민원창구] 등록 → 등록 완료 후 신고 수리
- 기술문서 포함내용 : 명칭(제품명, 품목명, 모델명) / 분류번호(등급) / 모양 및 구조 / 사용목적 / 사용방법 / 사용시 주의사항 / 제조원 (수입 또는 제조공정 위탁의 경우) / 동일성 제품 허가번호
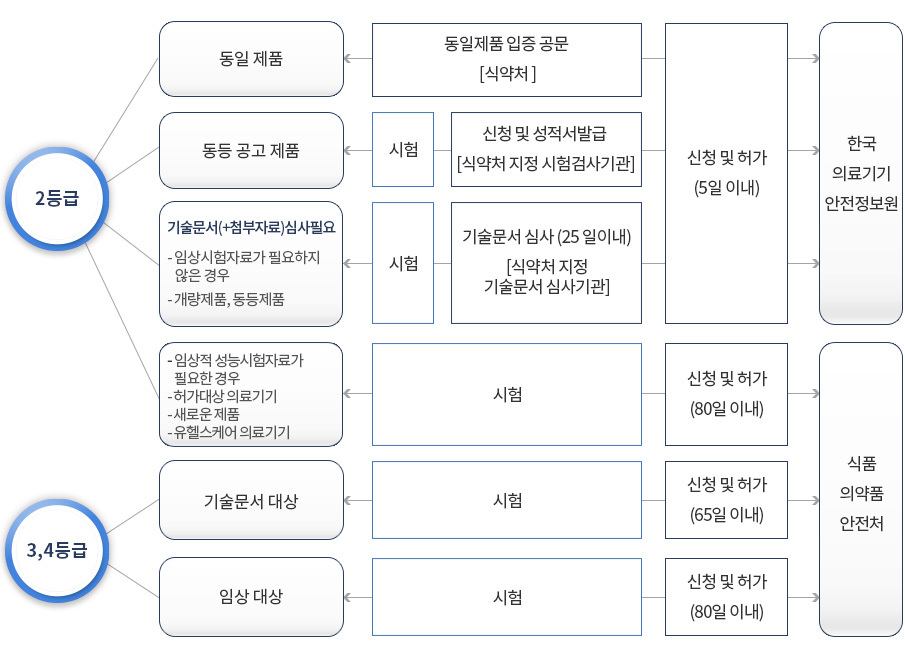
2등급 : 품목 인증(식품의약품안전처에서 진행)
- 품목분류 기준
- 동등제품 분류 기준 : 사용목적, 작용원리, 원재료, 성능
- 새로운 제품 : 이미 허가를 받은 체외진단 의료기기와 사용 목적, 작용 원리 등이 동등하지 아니한 체외진단 의료기기
- 개량제품 : 이미 허가를 받은 체외진단 의료기기와 시용 목적, 작용 원리는 동등하나, 원재료 또는 성능이 동등하지 아니한 체외진단 의료기기
- 동등제품 : 이미 허가를 받은 체외진단 의료기기와 시용 목적, 작용 원리, 원재료 및 성능이 동등한 체외진단 의료기기
3-4등급 : 품목 허가 (식품의약품안전처 본부에서 일괄 검토 및 진행)
- 2등급과 흐름은 동일. 기술문서 검토기간이 65일, 허가 일괄신청시 80일로, 기간에 차이가 있습니다.
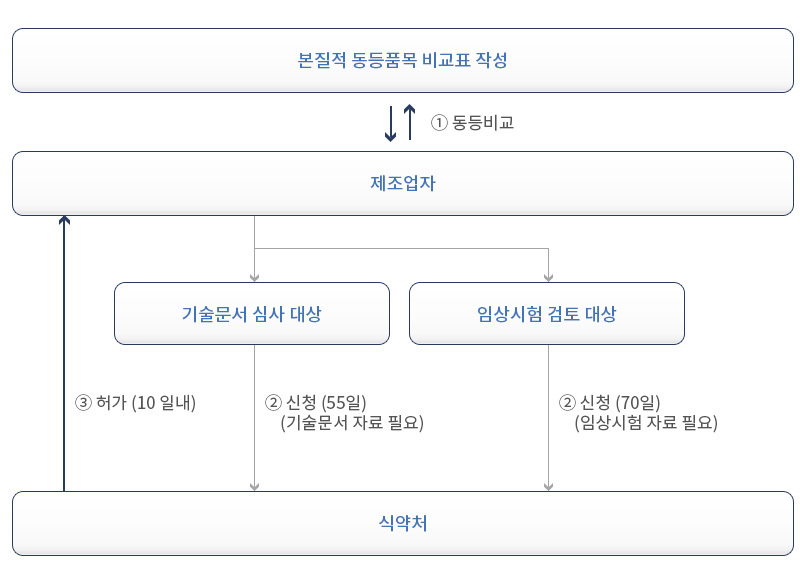
2등급 본질적 동등품목 비교대상 제품인증
한국(MFDS) 체외진단 의료기기 GMP
체외진단 의료기기GMP는 의료기기의 설계 · 개발, 생산, 설치 및 서비스를 제공함에 있어 적용되는 품질경영시스템의 요구사항을 규정한 것입니다.
GMP 세부 요구사항은 「체외진단 의료기기 제조 및 품질관리기준 고시」에서 확인할 수 있습니다.
모든 체외진단 의료기기 제조업자와 수입업자에게 적용되며, 심사 형태는 문서심사와 현장심사로 구분됩니다.
최초심사를 받은 후에는 3년마다 정기갱신심사를 받아야 합니다. GMP에서 분류하고 있는 품목군 중 기 인증받은 품목 외 추가품목이 발생할 경우, 소재지를 변경하는 경우에는 추가 현장심사가 있을 수 있습니다.
한국(MFDS) 의료기기 인,허가 컨설팅 추진단계


 10004 certified
10004 certified





